Chemissian 4.23
Chemissian 4.23 | 6.8 Mb
Chemissian is an analyzing tool of molecules electronic structure and spectra. It can manipulate molecular orbital energy-level diagrams (Hartree-Fock and Kohn-Sham orbitals), calculated and experimental UV-VIS electronic spectra, electronic/spin density maps and prepare them for publication. Chemissian has a user-friendly graphical interface and lets you examine and visualize data from the output of Gaussian, US-Gamess, Firefly/PC-Gamess quantum chemical program packages. Chemissian tools helps you to investigate nature of transitions in UV-vis spectra, bonding nature, etc.
Chemissian Features:
Build Molecular Orbitals energy level diagrams
• Due to the integrated graphical editor it is easy to add text labels to the diagrams, make connector lines between MO energy levels, text labels, occupy the energy levels with the electrons.
• You can analyze the electronic structure of molecules: you can move between energy levels simply using the keyboard cursor buttons and in a useful way obtain information about contributions to the current molecular orbital from atoms or molecular fragments and present the data in the most useful and demonstrative way: on the contribution diagram or directly on the MOs themselves
Build, visualize and interpret UV-Visible Spectra from Gamess, Firefly and Gaussian outputs
Chemissian with its exciting and unsurpassed graphical analyzer of properties and composition of MOs, has the wide range of capabilities for analysis of electronic spectra of molecules. Chemissian offer tools for building electronic UV/VIS spectra directly from quantum-chemical data from GAMESS, Firefly(PC-GAMESS) or GAUSSIAN output files:
• Build spectrum in one step: just load Gamess/Firefly/Gaussian outputs with TDDFT/CIS data.
• Having experimental spectra you can compare it with the calculated ones on the single diagram in the same wavelength scale.
• Any number of spectra may be added on single diagram, which is useful, e.g. when solvent influence on the spectrum is considered.
• Like in MOs editor it is possible to move between spectra peaks and correlate the current peak with transitions between MO energy levels.
• Chemissian allows editing the obtained spectrum diagram by adding text labels and other graphical objects. Different energy units are available.
• Using Chemissian tools it is easy to investigate the nature of spectra transitions, e.g. metal-to-ligand charge transfer, ligand-to-ligand charge transfer, pi-pi*, etc. based on information about the compositions of molecular orbitals from output files of gamess/Firefly/gaussian.
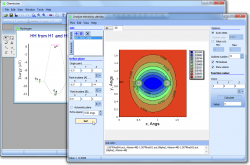
Analyze electronic density distribution
• Using Chemissian you can analyze electronic and spin density distribution, difference (also called "defomation") density, individual molecular orbital, and arbitrary linear combination of them (e.g. for plotting Fukui functions).
• Chemissian can build the distributions as two-dimensional contour maps or
• Build distribution along the given line (one-dimensional).
• To build densities only standart gamess/Firefly/gaussian output file is used, e.g. no cube-files are needed.
Calculate populations and valences
• Chemissian can calculate Mulliken and Simple populations of AOs, Shells, "Spherical Harmonics", Atoms or molecular fragments (any group of atoms).
• Also you can choose to calculate valences of AOs, Shells,"Spherical Harmonics", Atoms and fragments.
• Analyze molecular orbital composition - calculate contributions from atomic orbitals, atoms, molecular fragments, shells, etc. to the MOs.
Calculate quantum-chemical bond order indexes and overlap populations
Use Chemissian to investigate bonding nature in the molecules - calculate quantum-chemical bond order indexes and overlap populations for every bond in molecule. You can also analyze "generalized bond, e.g. "bond" between molecular fragments.
Work with several calculations at the same time
In a single document accumulate and analyze results of several calculations, e.g. load several GAMESS/Firefly/Gaussian ouput files. Simple example: having you the source reagents and the final reaction product you want to understand the changes that have occurred on the electronic structure level - you may add several calculations (reagent and product) at the same diagram, and they will be presented in the common energy scale, you can switch between different calculations, compare and analyze electronic structures all the participants at the same time.
Save the results in a single file
Save the obtained document in a special file format, which allows to keep all data in a single compressed file (uncompressed wave function takes up a lot of disk space!); at any time you will be able to open and continue working with the saved document, analyze, edit the data, send it to your colleagues.
Home Page - http://www.chemissian.com
Chemissian 4.23 | 6.8 Mb
Chemissian is an analyzing tool of molecules electronic structure and spectra. It can manipulate molecular orbital energy-level diagrams (Hartree-Fock and Kohn-Sham orbitals), calculated and experimental UV-VIS electronic spectra, electronic/spin density maps and prepare them for publication. Chemissian has a user-friendly graphical interface and lets you examine and visualize data from the output of Gaussian, US-Gamess, Firefly/PC-Gamess quantum chemical program packages. Chemissian tools helps you to investigate nature of transitions in UV-vis spectra, bonding nature, etc.
Chemissian Features:
Build Molecular Orbitals energy level diagrams
• Due to the integrated graphical editor it is easy to add text labels to the diagrams, make connector lines between MO energy levels, text labels, occupy the energy levels with the electrons.
• You can analyze the electronic structure of molecules: you can move between energy levels simply using the keyboard cursor buttons and in a useful way obtain information about contributions to the current molecular orbital from atoms or molecular fragments and present the data in the most useful and demonstrative way: on the contribution diagram or directly on the MOs themselves
Build, visualize and interpret UV-Visible Spectra from Gamess, Firefly and Gaussian outputs
Chemissian with its exciting and unsurpassed graphical analyzer of properties and composition of MOs, has the wide range of capabilities for analysis of electronic spectra of molecules. Chemissian offer tools for building electronic UV/VIS spectra directly from quantum-chemical data from GAMESS, Firefly(PC-GAMESS) or GAUSSIAN output files:
• Build spectrum in one step: just load Gamess/Firefly/Gaussian outputs with TDDFT/CIS data.
• Having experimental spectra you can compare it with the calculated ones on the single diagram in the same wavelength scale.
• Any number of spectra may be added on single diagram, which is useful, e.g. when solvent influence on the spectrum is considered.
• Like in MOs editor it is possible to move between spectra peaks and correlate the current peak with transitions between MO energy levels.
• Chemissian allows editing the obtained spectrum diagram by adding text labels and other graphical objects. Different energy units are available.
• Using Chemissian tools it is easy to investigate the nature of spectra transitions, e.g. metal-to-ligand charge transfer, ligand-to-ligand charge transfer, pi-pi*, etc. based on information about the compositions of molecular orbitals from output files of gamess/Firefly/gaussian.
Analyze electronic density distribution
• Using Chemissian you can analyze electronic and spin density distribution, difference (also called "defomation") density, individual molecular orbital, and arbitrary linear combination of them (e.g. for plotting Fukui functions).
• Chemissian can build the distributions as two-dimensional contour maps or
• Build distribution along the given line (one-dimensional).
• To build densities only standart gamess/Firefly/gaussian output file is used, e.g. no cube-files are needed.
Calculate populations and valences
• Chemissian can calculate Mulliken and Simple populations of AOs, Shells, "Spherical Harmonics", Atoms or molecular fragments (any group of atoms).
• Also you can choose to calculate valences of AOs, Shells,"Spherical Harmonics", Atoms and fragments.
• Analyze molecular orbital composition - calculate contributions from atomic orbitals, atoms, molecular fragments, shells, etc. to the MOs.
Calculate quantum-chemical bond order indexes and overlap populations
Use Chemissian to investigate bonding nature in the molecules - calculate quantum-chemical bond order indexes and overlap populations for every bond in molecule. You can also analyze "generalized bond, e.g. "bond" between molecular fragments.
Work with several calculations at the same time
In a single document accumulate and analyze results of several calculations, e.g. load several GAMESS/Firefly/Gaussian ouput files. Simple example: having you the source reagents and the final reaction product you want to understand the changes that have occurred on the electronic structure level - you may add several calculations (reagent and product) at the same diagram, and they will be presented in the common energy scale, you can switch between different calculations, compare and analyze electronic structures all the participants at the same time.
Save the results in a single file
Save the obtained document in a special file format, which allows to keep all data in a single compressed file (uncompressed wave function takes up a lot of disk space!); at any time you will be able to open and continue working with the saved document, analyze, edit the data, send it to your colleagues.
Home Page - http://www.chemissian.com
التعديل الأخير بواسطة المشرف:
")